Rarefaction - Is it a good idea?
Today, I came across an Editorial from the journal Microbiome - here - on concepts and recommendations in microbiota studies, including data analysis. To my surprise, I found the following sentence:
“Rarefaction techniques are recommended to be used for specific analyses that are known to be sensitive to sequencing depth”.
It is baffling to me to see this as a recommendation, considering the evidences of the problems it entails. Why do I say this?
What is rarefaction
Rarefaction is a frequent approach in microbiome studies. It consists on subsetting the samples of a study to a threshold of reads, in order to normalise the data and remove the effect of uneven sequencing depth. This is considered to be essential, since variation of sequencing depth is stochastic and can lead to biased results.
Let M1 and M2 be two different microbiome samples. If the sequencing depth from M1 is higher than M2, then richness (an alpha-diversity metric) will be higher in M1 than in M2, even if there is no real difference between the two samples. To address this, M1 is then rarefied, or the reads of M1 are subsetted in order to create now an M1’ with the same sequencing depth as M2.
It seems to be a straighforward method to account for sequencing depth, so why all the fuss about it?
More than just meets the eye - what are the concerns with rarefaction?
In short, rarefaction is just throwing away good data. By doing so, we are losing information about our samples and biasing our metric estimates - if nothing else, we are getting very biased estimates on the variance. The consequences of rarefaction are very well illustrated in the amazing paper by Paul McMurdie and Susan Holmes called Waste not, want not: why rarefying microbiome data is inadmissible.
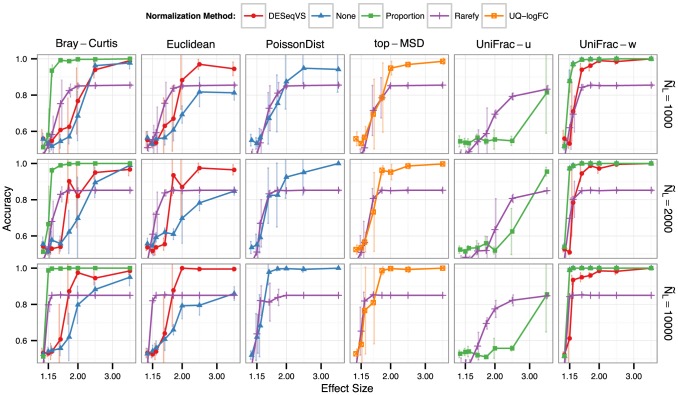
The authors demonstrate that rarefaction affects both diversity metrics, as well as abundance estimates.
Starting with diversity metrics, the authors show that rarefaction leads to less accurate results in several distance metrics in comparison with other methods, ones more simple (proportion) than others (DESeq2 variance stabilization). The only one where rarefaction outperforms proportion in is the unweighted Unifrac distance. However, this is not a surprise, since unweighted unifrac, which focus on presence/absence data, is easily influencied by the sequecing depth. Nevertheless, it is also important to state this metric can also contain noise, since it cannot separate true rare lineages from noise (introduced by singletons, for example).
Regarding abundance, the insult is even bigger. First of all, even with rarefaction, traditional two-group t-tests show a poor performance when comparing abundance between groups - this stems from the fact microbiome data needs robust statistical methods to deal with its nature - this topic can be left for a different post. Nevertheless, what it is shown here is that even solid statistical models, like the one implemented in DESeq2, show worst results when the samples are previously rarefied versus the raw samples.
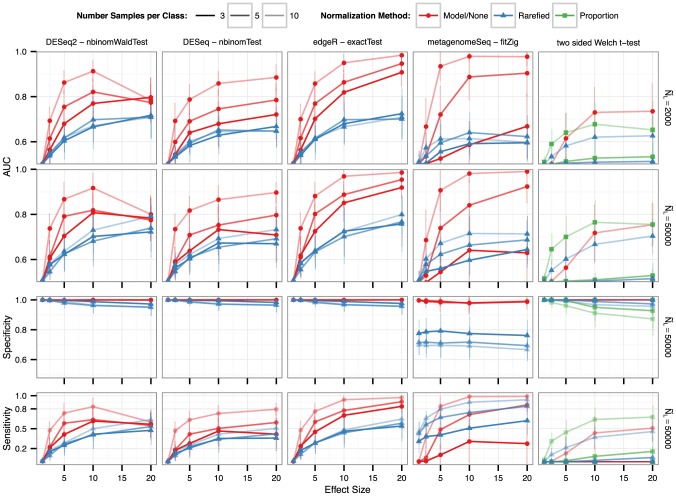
For additional results and to fully understand the issues that rarefaction causes, I really recommend reading the paper from McMurdie & Holmes.
Ok ok, rarefaction is bad. So… what should I do?
I understand how complicated is to navigate in the literature concerning microbiome methodology, especially with so many tools, approaches and opinions. However, in my view, looking at methods that can account for differences in sequencing depth across samples, ranging from simple proportions to more complex ones, such as the variance stabilizing transformation implemented in DESeq2, is useful. Using abundance-based metrics with such approaches for beta-diversity or introducing estimates on unobserved species, as suggested here by Amy D. Willis, can yield more accurate results than rarefaction.
Regarding abundance, several count-based models such as the ones for RNASeq - microbiome data is reads per ASV, which is the same structure as transcriptomic data - are frequently used. Other methods such as Dirichlet regressions or ANCOM II address several issues, such as sequencing depth but also the compositional nature of the data (again, a topic for a different post).
In any case, if you have any question, feel free to contact your local statistician (such as the Data Misfits team)!